
Ultrafast Spectroscopy and Excited-State Dynamics
The accurate measurement and simulation of the dynamics of ultrafast processes such as excited-state intramolecular proton transfer (ESIPT) or photoisomerization in the polyatomic molecules has been the focus of much recent nterest. Transient absorption (TA) spectroscopy is a promising technique to measure the dynamics of these ultrafast process due to its excellent time resolution and applicability to a diverse class of systems. However, the spectral “blobs” observed in these ultrafast techniques are usually less informative regarding the underlying dynamics due to the involvement of large number of active degrees of freedom which are projected onto an observable with fewer dimensions. Global analysis of ultrafast spectra may provide kinetic time constants from the decay of signals, etc., but still leave much room for the comprehensive assignment of the components of the spectrum and extraction of relevant physical quantities. This state-of-the-art suggests that it is critical to compare the experimental ultrafast signals with ab initio theory that directly simulates the experiment and the relevant observables.
First-principles nonadiabatic molecular dynamics methods, such as ab initio multiple spawning (AIMS) provide a powerful toolkit to interpret the ultrafast experiments without any empirical inputs. I developed a methodology combining the AIMS simulations with the GPU-accelerated Time-dependent Complete Active Space Configuration Interaction (TDCASCI) method to simulate the excited state dynamics and gas-phase TA spectrum of molecules and nanomaterials. My developed protocol calculates the TA spectrum using the representative geometries (green circles in the illustration below) from the trajectories of AIMS simulations (dotted brown lines) separated by a weighted k-means clustering algorithm. For each time-separated snapshot, the stimulated emission and excited state absorption components are computed by using electric dipole approximation applying a δ-function pulse polarized separately along the x, y and z directions of the molecular axis to the initial electronic state. The simulation of the TA spectrum involves the running thousands of individual TD-CASCI simulations, which highlights the power of GPU-accelerated implementations of the method that uses a direct configuration interaction approach to eliminate the need for explicitly building or diagonalizing the Hamiltonian matrix.
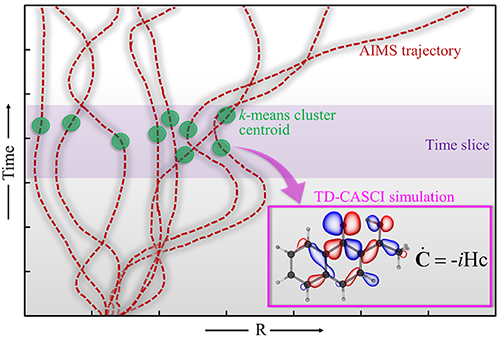
The initial application of my approach on prototypical molecules Salicylideneaniline, 1’-Hydroxy-2’-acetonaphthone and 2-Nitrophenol showed that the simulated spectra are highly consistent with the gas-phase experimental TA studies. My methodology provides the added advantage of separately simulating the spectrum of excited state trajectories evolved on the ground vs excited state and/or those undergoing distinct photochemical relaxation pathways, providing a more comprehensive and critical insight into the experimental TA spectrum. Additionally, the methodology provides a dynamic picture of TA spectrum evolution along a specific degree of freedom like a movie (see illustration below), paving the way towards better assignment of the components of the experimental spectrum.
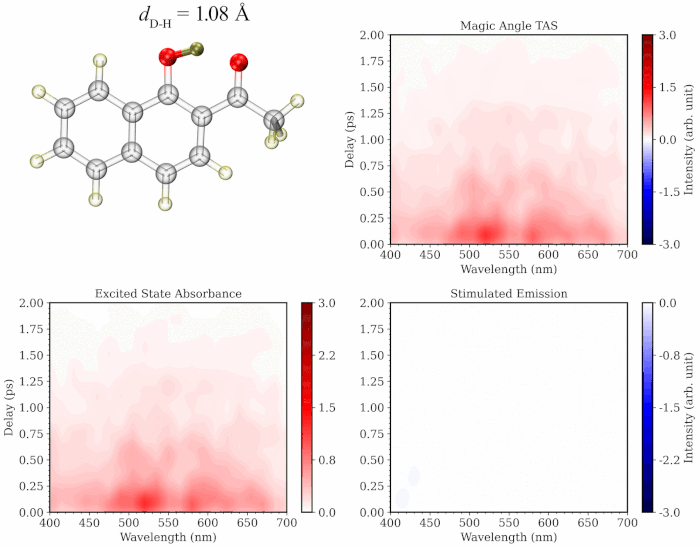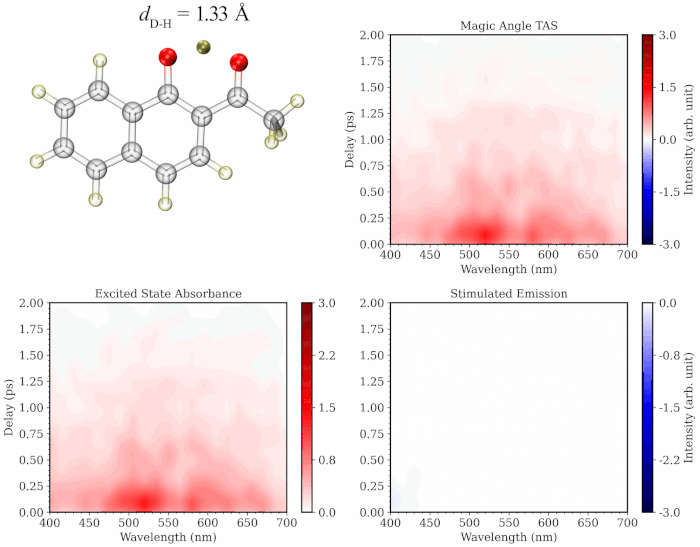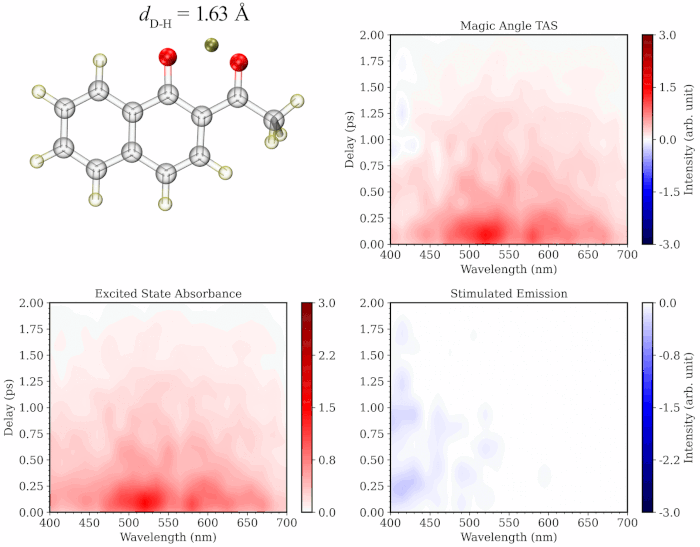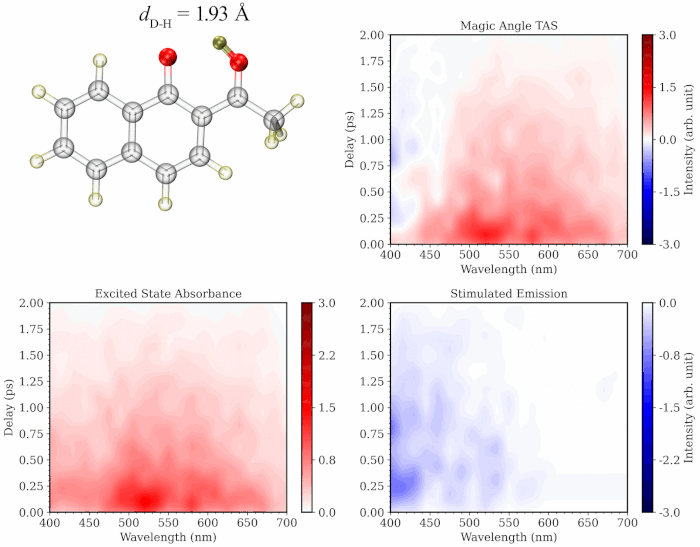
To learn more about this topic, please refer to my papers on Salicylideneaniline and 1’-Hydroxy-2’-acetonaphthone, as well as this poster.