
Quantum Crystallography
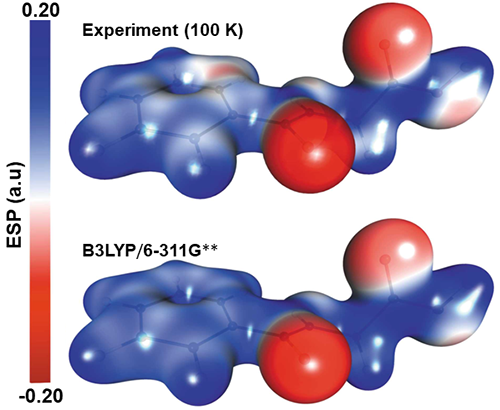
I have deep interests in experimental and theoretical charge density analysis of small molecules. The refinement of experimental X-ray and electron diffraction data using Hansen-Coppens multipolar model offers highly efficient tools to extract precise information about the charge density distributions on the molecules. The obtained electron density can be analyzed in the realm of “Quantum Theory of Atoms in Molecules (QTAIM)” to characterize the nature of covalent and non-covalent interactions in terms of electron density, \(\rho(\overrightarrow{\mathrm{r}})\), gradient vector field of the electron density, \(\nabla \rho(\overrightarrow{\mathrm{r}})\), and Laplacian of the electron density, \( \nabla^2 \rho(\vec{r})\), associated with the bond critical point (BCP). DFT based theoretical studies are extensively used to measure the correctness of structure factors extracted from experimental measurements and to complement the experiment QTAIM based bonding descriptors.
I have developed a Python-based program that performs Fourier transforms of electron densities (both valence and core) obtained from periodic Density Functional Theory (DFT) calculations using software like Quantum-ESPRESSO or CP2K. This allows us to derive static theoretical structure factors in crystalline phases up to the experimental \( \sin \theta / \lambda \) limits. Previously, such capabilities were primarily found in commercial programs such as CRYSTAL, WIEN2k, and VASP. The resulting structure factors can be directly applied for multipolar refinement of charge density parameters using packages such as MOPRO or XD, complementing results obtained from experimental diffraction studies (see illustration below).
For more details on these projects, refer to articles 10, 13, 15, 16, 19, 20, 22, 23, and 24 from my publications.